For US healthcare professionals only
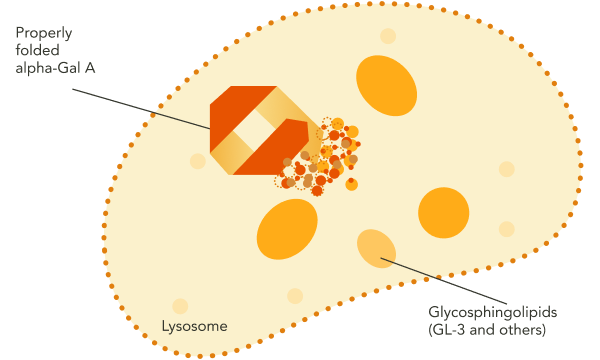
GLA encodes the lysosomal enzyme alpha-galactosidase A (alpha-Gal A).1 In individuals without Fabry disease, functional, correctly folded alpha-Gal A is trafficked from the endoplasmic reticulum, where it is synthesized, to the lysosome, where it breaks down certain glycosphingolipid substrates, including GL‑3 and lyso‑Gb3.1-3
In Fabry disease, the absence of functional alpha-Gal A results in accumulation of GL‑3 and other disease-causing substrates in the lysosome.1 This substrate accumulation triggers a cascade of pathogenic processes.1 Over time, the resulting organ damage may become irreversible.1
Fabry disease can be caused by a variety of missense or nonsense point mutations, splicing mutations, small deletions or insertions, and large deletions.1 Some of these mutations render alpha-Gal A absent or grossly affect its structure, while others, often affecting a single amino acid, result in an intact but misfolded protein that is unable to be trafficked to the lysosome.1,3,4

These disease processes may result in signs and symptoms of Fabry disease across multiple organ systems.1
GL-3, globotriaosylceramide; GLA, galactosidase alpha gene; lyso-Gb3, globotriaosylsphingosine.
References: 1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. 2. Parenti G, Andria G, Valenzano KJ. Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol Ther. 2015;23(7):1138-1148. 3. Brooks DA. Getting into the fold. Nat Chem Biol. 2007;3(2):84-85. 4. Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19(4):430-438.